| Key inclusion criteria |
- Histologically or cytologically confirmed stage III (non-N3) NSCLC (TNM 8th edition), before start of concurrent CRT.
- In case of suspected N2-disease, mediastinal invasive staging (EUS, EBUS and/or mediastinoscopy) to prove N2 involvement before start of concurrent CRT is preferred.
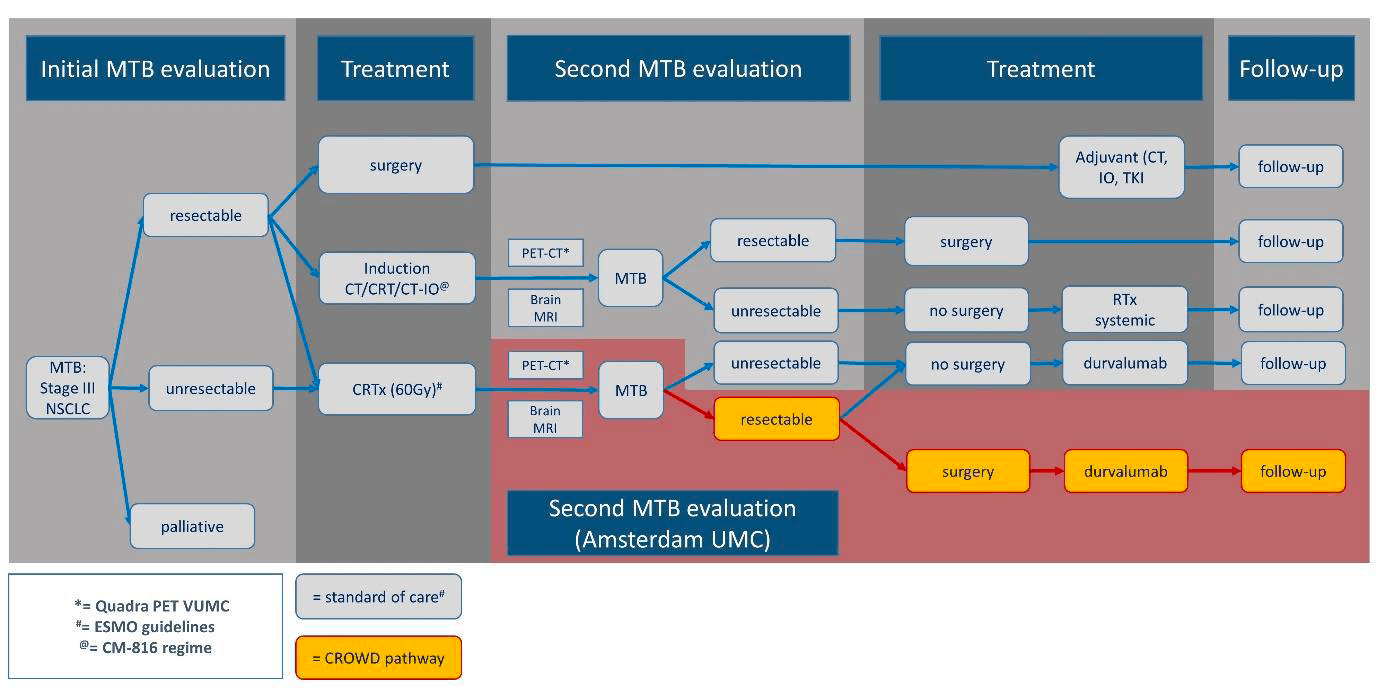
- Initial MDT recommendation for a non-surgical treatment comprising concurrent CRT (platinum-doublet, 60 Gy in 30 fractions of 2 Gy once daily) followed by durvalumab
- Capable of giving signed informed consent which includes compliance with the requirements and restrictions listed in the informed consent form (ICF) and in this protocol. Written informed consent and any locally required authorization (eg, European Union [EU] Data Privacy Directive in the EU) obtained from the patient/legal representative prior to performing any protocol-related procedures, including screening evaluations.
- Age > 18 years at time of study entry, i.e. the day of signing informed consent.
- Have a performance status of 0-1 on the ECOG Performance Scale at time of restaging.
- Body weight >30 kg
- Demonstrate adequate normal organ and marrow function, as deemed acceptable by the treating physician in the context of CRT:
- Leukocytes ≥ 3,000/mm3
- Absolute neutrophil count (ANC) ≥ 1,000/mm3
- Platelet count ≥ 75,000/mm3
- Hemoglobin ≥ 6 mmol/L (9.7 g/dL)
- Creatinine ≤ 1.5 x ULN or creatinine clearance (CrCl) ≥40 mL/min (if using the Cockcroft-Gault formula below): Female CrCl = [(140 - age) x weight x 0.85]/(0.85 x creat in mmol/L) Male CrCl = [(140 - age) x weight x 1.00]/(0.81 x creat in mmol/L)Total Bilirubin ≤ 1.5 x ULN (except subjects with Gilbert Syndrome)
- AST and ALT ≤ 3 times the upper limit of normal
- Subjects must have adequate lung function to permit surgical resection determined by pre-enrollment pulmonary function tests to include DLCO
- Patient is willing and able to comply with the protocol for the duration of the study including undergoing treatment and scheduled visits and examinations including follow up.
|
| Key exclusion criteria |
- Patients with TxN3 or M1 disease
- Patients with known actionable genomic alterations.
- The use of any CRT schemes other than those in ESMO recommendations (platinum doublet, 30 once-daily fractions of 2 Gy)
- Patients deemed inoperable based on cardiopulmonary function tests or comorbidity
- Unable to undergo CT-scan with iv-contrast
- Unable to remain supine for 15 min for the PET-low-dose CT scan
- Has received prior therapy with an anti-PD-1, anti-PD-L1, anti-PD-L2, anti-CTLA-4 antibody, or any other antibody or drug specifically targeting T-cell co-stimulation or immune checkpoint pathways.
- Participation in another clinical study with an investigational product during the last 4 weeks.
- Concurrent enrolment in another clinical study, unless it is an observational (non-interventional) clinical study or during the follow-up period of an interventional study
- Any unresolved toxicity NCI CTCAE Grade ≥2 from previous anticancer therapy with the exception of alopecia, vitiligo, and the laboratory values defined in the inclusion criteria
a. Patients with Grade ≥2 neuropathy will be evaluated on a case-by-case basis after consultation with the Study Physician.
b. Patients with irreversible toxicity not reasonably expected to be exacerbated by treatment with durvalumab may be included only after consultation with the Study Physician.
- Receipt of the last dose of anticancer therapy (chemotherapy, immunotherapy, endocrine therapy, targeted therapy, biologic therapy, tumor embolization, monoclonal antibodies) ≤4 weeks prior to the first dose of study drug. If sufficient wash-out time has not occurred due to the schedule or PK properties of an agent, a longer wash-out period will be required, as agreed by AstraZeneca/MedImmune and the investigator.
- Any concurrent chemotherapy, IP, biologic, or hormonal therapy for cancer treatment. Concurrent use of hormonal therapy for non-cancer-related conditions (e.g., hormone replacement therapy) is acceptable.
- Major surgical procedure (as defined by the Investigator) within 28 days prior to the first dose of IP.
- History of allogenic organ transplantation.
- Active or prior documented autoimmune or inflammatory disorders (including inflammatory bowel disease [e.g., colitis or Crohn's disease], diverticulitis [with the exception of diverticulosis], systemic lupus erythematosus, Sarcoidosis syndrome, or Wegener syndrome [granulomatosis with polyangiitis, Graves' disease, rheumatoid arthritis, hypophysitis, uveitis, etc]). The following are exceptions to this criterion:
a. Patients with vitiligo or alopecia
b. Patients with hypothyroidism (e.g., following Hashimoto syndrome) stable on hormone replacement
c. Any chronic skin condition that does not require systemic therapy
d. Patients without active disease in the last 5 years may be included but only after consultation with the study physician
e. Patients with celiac disease controlled by diet alone
- Uncontrolled intercurrent illness, including but not limited to, ongoing or active infection, symptomatic congestive heart failure, uncontrolled hypertension, unstable angina pectoris, cardiac arrhythmia, interstitial lung disease, serious chronic gastrointestinal conditions associated with diarrhea, or psychiatric illness/social situations that would limit compliance with study requirement, substantially increase risk of incurring AEs or compromise the ability of the patient to give written informed consent
- History of another primary malignancy except for
a. Malignancy treated with curative intent and with no known active disease ≥5 years before the first dose of IP and of low potential risk for recurrence
b. Adequately treated non-melanoma skin cancer or lentigo maligna without evidence of disease
c. Adequately treated carcinoma in situ without evidence of disease
- History of leptomeningeal carcinomatosis
- History of active primary immunodeficiency
- Known active hepatitis infection, positive hepatitis C virus (HCV) antibody, hepatitis B virus (HBV) surface antigen (HBsAg) or HBV core antibody (anti-HBc), at screening. Participants with a past or resolved HBV infection (defined as the presence of anti-HBc and absence of HBsAg) are eligible. Participants positive for HCV antibody are eligible only if polymerase chain reaction is negative for HCV RNA. Adjust wording as necessary and consider evaluating at screening for studies with known hepatotoxicity or other relevant requirements.
a. Participants co-infected with HBV and HCV, or co-infected with HBV and HDV, namely: HBV positive (presence of HBsAg and/or anti HBcAb with detectable HBV DNA); AND
b. HCV positive (presence of anti-HCV antibodies); OR
c. HDV positive (presence of anti-HDV antibodies).
- Known to have tested positive for human immunodeficiency virus (HIV) (positive HIV 1/2 antibodies) or active tuberculosis infection (clinical evaluation that may include clinical history, physical examination and radiographic findings, or tuberculosis testing in line with local practice).
- Current or prior use of immunosuppressive medication within 14 days before the first dose of durvalumab. The following are exceptions to this criterion:
a. Intranasal, inhaled, topical steroids, or local steroid injections (e.g., intra articular injection)
b. Systemic corticosteroids at physiologic doses not to exceed 10 mg/day of prednisone or its equivalent
c. Steroids as premedication for hypersensitivity reactions (e.g., CT scan premedication)
- Receipt of live attenuated vaccine within 30 days prior to the first dose of IP. Note: Patients, if enrolled, should not receive live vaccine whilst receiving IP and up to 90days after the last dose of IP.
- Female patients who are pregnant or breastfeeding or male or female patients of reproductive potential who are not willing to employ effective birth control from screening to 90 days after the last dose of durvalumab monotherapy.
- Known allergy or hypersensitivity to any of the study drugs or any of the study drug excipients.
Prior randomization or treatment in a previous durvalumab clinical study regardless of treatment arm assignment.
- Judgment by the investigator that the patient is unsuitable to participate in the study and the patient is unlikely to comply with study procedures, restrictions and requirements.
|